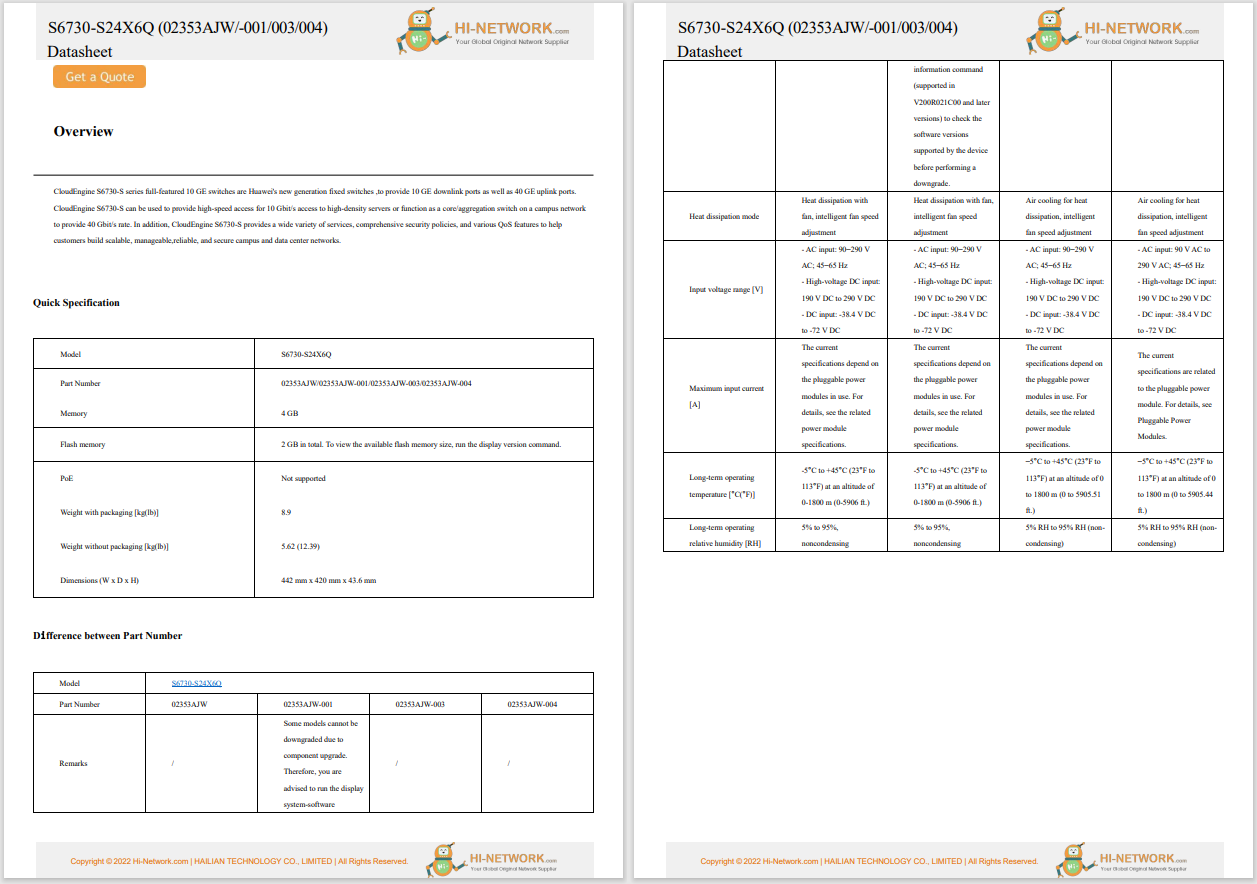
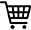Cadastre-se agora para um orçamento mais personalizado!

















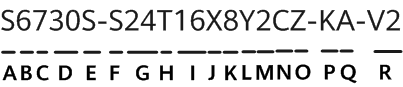











The breakthrough of CRISPR technology in the past two decades has allowed biologists to refine the manipulation of DNA, to slice and dice it in order to create organisms tailored to particular purposes. That free-wheeling editing of genes, though, produces a new problem: how to organize all the complexity of the different edited pieces of DNA.
That's especially important for the multi-hundred-billion-dollar portion of the drug market called biologics, basically engineered proteins that can achieve a particular purpose. If you're going to engineer new proteins through CRISPR, you need to do it in a systemic way, which is a fairly demanding combinatorial problem.
Hence, some smart young biotechs are turning to deep learning forms of artificial intelligence, as deep learning is a technology that loves combinatorial problems.
Biotech firm Absci, which came public last year, was founded a decade ago by CEO Sean McClain, who came up with a novel way to engineerE. colicells as factories for producing custom proteins that a drug maker would want, such as monoclonal antibodies that can fight viruses. You could say McClain is the Elon Musk of protein manufacturing.
Greater manufacturing capability engendered a new problem: What to make, exactly.
Shortly before going public, Absci bought another startup, Denovium, a three-year-old firm pioneering deep learning to analyze all the many combinations of proteins that McClain's cells can churn out.
"We've built a very large library of these genetic parts, and we can snap them together combinatorially," says Absci chief technologist Matthew Weinstock. "And which sequence of DNA is best to produce this protein is the problem of codon optimization, and it's a very big challenge."
Absci Inc."We've built a very large library of these genetic parts, and we can snap them together combinatorially," explained Absci chief technologist Matthew Weinstock in a meeting withZDNetvia Zoom. "And which sequence of DNA is best to produce this protein is the problem of codon optimization, and it's a very big challenge."
"If we have a million to a billion different cell lines, we need a screening capability that allows us to go through them to fish out the needles from the haystack, to find these genetic designs are the right ones."
Not only is the manufacture of proteins a combinatorial challenge, but so is the determination of which protein will work as a biologic for a given disease, the fundamental question of drug discovery.
"We can randomize the protein sequence itself and ask what protein sequence is the best for binding to this particular target," said Weinstock.
Weinstock, who has a PhD in biochemistry from the University of Utah, had previously run the development of next-gen therapeutics at startup Synthetic Genomics, Inc. There, he met up with Gregory J. Hannum, a PhD in bioengineering from UC San Diego. Hannum would go on to found Denovium in order to build deep learning tools.
Following the acquisition a year ago, Hannum became co-lead of AI research at Absci, along with his Denovium co-founder, Ariel Schwartz.
"Biology is one of the most complex problems that the planet has," said Hannum in the same interview withZDNet.
"It's essentially a self-bootstrapped system, billions of years in the making that, if we could just understand what all the different letters are, and what their combinations were, we'd have tremendous power to engineer new drugs and help humanity in new ways."
The field of biology has built "beautiful databases" by wet-lab observation, notes Hannum, such as the UniProt database or Universal Protein Resource, which is maintained by a consortium of research centers around the world, and which is funded by a gaggle of government offices, including the U.S.'s National Institutes of Health and National Science Foundation.
Despite those beautiful databases, and despite basic analysis with techniques such as Hidden Markov Models, a third of all proteins remain a mystery in terms of their function.
To try and resolve the mystery, Denovium built one giant model to tackle all proteins at once.
"Rather than have hundreds of thousands of small models, we built one deep learning model that can go straight from sequence to function."
That giant model has what's called an "embedding," a representation of proteins that is "very generalizability," said Hannum. Think of it as compressing what is known about the protein down to a set of points that would reproduce what's known about any protein.
See also:Google DeepMind's effort on COVID-19 coronavirus rests on the shoulders of giants.
"This gives us a ton of advantages," said Hannum. "We can annotate proteins," meaning, assign hypotheses about their functions, "a lot of which had never been understood."
In addition, it can find novel proteins whose amino acid sequence is still unknown by finding functional homologues that have similar properties to the known ones.
The model can also make predictions as to what changing amino acid sequences might do. "You know this has DNA-binding properties; what if I change this base," meaning amino acid-base, said Hannum.
"Scientists took decades to build Uniprot," he observed. With the Denovium model, Absci can re-run its predictions against the Uniprot database during a weekend. "We can generate tremendous new information."
Denovium didn't just study proteins; it also built a program called Gateway to connect DNA and proteins. Gateway links DNA and protein representations in one model to let a scientist "drag and drop a whole genome, and find every protein, and annotate their functions, all in a single model, which is still state of the art," said Hannum.
Once inside Absci, the challenge for Hannum and Schwartz moved from just annotating DNA and proteins to solving the manufacturing problem that Weinstock was dealing with.
One example is finding novel "chaperones," proteins that guide the folding of proteins. "We can take the ones we knew about, and find many, many more" by sensing similarities between known and unknown, said Hannum. "Rather than just finding a list of them, we can actually characterize them into functional groups, say these are similar, and build a whole map of all the proteins related to how they help other proteins fold."
That function is "really unique," said Weinstock. It has boosted Absci's production of proteins more than two-fold.
The right chaperone protein, in this case, is not one anyone would have thought would work when regarded with traditional bioinformatics tools. "It was a protein of unknown function, from an obscure root bacterium," said Weinstock. "But the model actually told us this is probably a chaperone, and it led us to give it a try."
To build the giant model at Denovium, Hannum and Schwartz began with what he called "rather primitive" approaches, using convolutional neural networks, or CNNs, the workhorse of image recognition.
Since those early efforts, the team embraced Transformers, the large attention-based models developed at Google, and "a lot of the architectures around there." There are many ways, he said, that natural language processing of the sort done by Transformers can complement image recognition.
That has echoes to DeepMind's protein-folding program AlphaFold, which in its second version, this past summer, moved from using convolutions to using attention-based models.
See also: DeepMind's AlphaFold 2 reveal: Convolutions are out, attention is in.
"NLP fields and vision fields have gone their own separate ways, but I feel there's more that can be learned from each side," said Hannum. "We're really looking to combine the best of both worlds."
According to Hannum, the ideal representation of proteins is "something we're currently evaluating, where to go." The original form of representation "was flat; it's an affine," he said, meaning an abstract form that simply says multiple things are related so as to be as broad as possible.
"The intention of that [Denovium] engine was to create a very unstructured representation because it then had the flexibility to contain arbitrary context -- structure, homology, everything -- it contains everything in vector space." Another way of describing it, he said, is "a not-quite-Gaussian distribution, point-cloud of every protein in the protein universe."
"Biology is one of the most complex problems that the planet has," says Absci's co-lead for A.I., Gregory J Hannum. "It's essentially a self-bootstrapped system, billions of years in the making that, if we could just understand what all the different letters are, and what their combinations were, we'd have tremendous power to engineer new drugs and help humanity in new ways."
Absci Inc.(If you want to get a more-specific sense of that representation, you can check out a paper written by Hannum and Schwartz when they were at Synthetic Genomics and posted on BioRxiv, describing something called "Deep Semantic Protein Annotation Classification and Exploration.")
What, one wonders, would be theloss function, the way that the Denovium engine gets better with its broad function. "Our loss function is multitudes," said Hannum, "it's highly multi-task; we believe that's key to generalization."
That includes learning labels of how proteins fold in a given region. But it also learns sequence clustering groups to detect homology between proteins. There are "letter-specific" outputs, he said, that tells Absci if, given a certain amino acid-base, a protein might be doing something such as functioning as a membrane protein of a cell.
Each of those different kinds of tasks can dominate at any point in research, given Absci has a complex pipeline to solve. So, the larger question becomes, What is the reward signal for Absci? What tells the company it will make real progress in developing cell lines that develop the right biologic.
What is optimal rather than simply progress, in other words?
"It's a fantastic question," said Hannum, "and it's one that is something we are very passionate about demonstrating."
He is inclined to view the goal as narrowing the search space to advance the drug discovery task overall. That means, "How good were your starting points? Can we narrow that list down?"
For example, "It's nice to get one new chaperone that made a contribution to a project, but if we could say, next time, try these chaperones, and one of them is going to be that new winner, you can see the continual improvement over time with that."
The combinatorial problem for a given antibody, across its 60-odd residues, is more possibilities than there are atoms in the known universe, notes Weinstock. In his view, the value of Denovium's engine is to find those things in both manufacturing of cell lines and in drug discovery that wouldn't have been considered in the wet lab.
"These technologies are going to allow us to extend to solutions that didn't even exist in the original cell library," he said, "being able to say, This antibody sequence is most optimal -- you didn't even test it, but the model is able to predict it, or this is the best cell line, or this is the best ribosome binding site."
The end game is to move to anin-silicoapproach for most if not all of Absci's work, where less and less has to be done with the tedium of the wet lab.
"The vision is to have something that straight-up solves it," said Hannum. "You ask it, I have to treat this, then the computer designs the antibody and the manufacturing solution, and in a very short time, you're already throwing the solution in the tank and making your drug."
As Weinstock sees it, "If we can eliminate that few months of work in the secondary screening campaign" in the wet lab, "and turn it into a computational problem we can solve over the weekend, not only are we going to get better outputs, but we're going to get those faster."
As proof of progress so far, Weinstock and Hannum point to the ability to move beyond published deep learning approaches in the development of biologics. For example, a study published last year inNature Biomedical Engineeringby scientists at deepCDR Biologics, and ETH Zurich university, in Zurich used deep learning to solve the problem of whether or not certain antibodies will bind to their targets on a disease, the antigen.
But, Absci claims in an internal case study, its deep learning is able to predict not just a yes or no answer for binding of an antibody but also quantify thedegreeof binding "affinity."
Said the company, in an email toZDNet:
Going beyond that "state-of-the-art," Absci's model (along with its proprietary wet lab assays) demonstrates high performance quantitative predictions about the affinity of antibody/antigen interaction. Measured as "Kd" - the dissociation constant - this is essentially a measure of how well the antibody sticks to its antigen, and it is a critical determinant of drug functionality. Thus the model allows Absci to design in silico new antibodies with desired target binding affinity.
The Absci approach is continuing to win converts. As of September, the company had 9 of what it calls "Active Programs," those where the company has "negotiated, or expect to negotiate, license agreements for downstream milestones and royalties" with various clients.
Subsequently, in October, it signed a multi-program discovery deal with Cambridge, Mass-based pharma EQRx. And Friday, the company announced it has formed a research collaboration with drug giant Merck to design "to produce enzymes tailored to Merck's biomanufacturing applications." The agreement includes the prospect for Merck to eventually "nominate up to three targets for drug discovery," the two firms said.
That announcement drove up the stock by 26% Friday.
Absci toldZDNetit will be updating its total number of Active Programs "in future disclosures." The company will be presenting at J.P. Morgan's healthcare conference on Monday, January 10th, at 12:45 pm, Pacific time, and you can live stream it.
Nevertheless, the world is still waiting for that day when it can be declared that A.I. has led to a drug that was never before possible, Weinstock concedes.
"In terms of seeing a molecule in the clinic that was designed with some of these technologies, I think we're probably two years out," said Weinstock.
 Tags quentes :
Inteligência artificial
Inovação
Tags quentes :
Inteligência artificial
Inovação
Cadastre-se agora para Ações Semanais de Promoção
100% free, Unsubscribe any time!
Add 1: Room 605 6/F FA YUEN Commercial Building, 75-77 FA YUEN Street, Mongkok KL, HongKong Add 2: Room 405, Building E, MeiDu Building, Gong Shu District, Hangzhou City, Zhejiang Province, China
Whatsapp/Tel: +8618057156223 Tel: 0086 571 86729517 Tel em HK: 00852 66181601
Email: [email protected]
 English
English Pусский
Pусский Français
Français Español
Español Português
Português